
Curare le malattie rare: l’esperienza di Federico, alle prese con la RTD
| di Federico Autunno

In occasione della giornata di sensibilizzazione organizzata dalla fondazione CURE RTD, ho deciso di metter sù quattro righe che parlassero della mia esperienza personale con questa malattia, e si ponessero come obiettivo l’idea di avvicinare quante più persone possibili al mondo delle malattie rare, in questo caso della RTD.
Il mondo delle malattie rare è qualcosa di paradossale e misterioso per la maggioranza delle persone, una malattia è rara nel momento in cui presenta una casistica statisticamente marginale all’interno della società. Esistono innumerevoli tipologie di malattie rare, alcune più rare ed altre meno rare. In questo articolo vorrei parlare della mia esperienza con una patologia, non rara, ma rarissima: la “Brown-Vialetto-Van Laere”, poi rinominata nella moderna RTD, dall’inglese “Riboflavin Transporter Deficiency” (carenza del trasportatore di riboflavina, ossia la vitamina B2).
Si tratta di una patologia rarissima, tanto che la sua presenza nella popolazione mondiale è stimata 1/milione. La RTD è dovuta alle mutazioni di tre geni che, a seconda dei casi, sono, rispettivamente, responsabili delle tre diverse forme con cui la malattia può presentarsi. Esistono, infatti, tre tipi di RTD: di tipo 1 se le mutazioni coinvolgono il gene SLC52A1, ma le più comuni sono di tipo 2 se le mutazioni riguardano il gene SLC52A2, e di tipo 3 nel caso in cui il gene coinvolto sia SLC52A3. Tutti questi geni sono responsabili della RTD in quanto svolgono il fondamentale ruolo di fornire istruzioni per produrre le proteine trasportatrici delle riboflavina (vitamina B2).
Queste tre forme della malattia differiscono tra loro per la variazione di alcuni sintomi: la RTD di tipo 1 è quasi assente nella casistica di questa patologia (un solo caso riportato), nel caso della RTD di tipo 2 e di tipo 3, sebbene vi siano molti sintomi sovrapponibili, entrambe presentano fenotipi distinti. La RTD di tipo 2, ad esempio, è caratterizzata da una debolezza muscolare prevalentemente negli arti superiori e dalla perdita della vista dovuta all’atrofia ottica, invece, nel caso dell’RTD di tipo 3 la debolezza muscolare è più generalizzata su tutto il corpo.
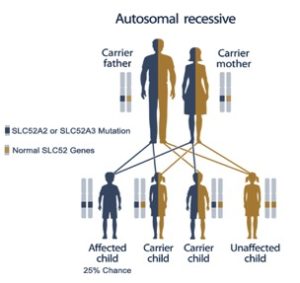
Le mutazioni genetiche alla base dell’RTD sono anche il motivo per cui questa patologia è così rara. Stiamo parlando di una patologia la cui trasmissione avviene in modo autosomico recessivo, nel senso che gli individui con RTD hanno due copie mutate del gene responsabile della malattia. Dunque, un individuo con RTD non può che essere il frutto dell’unione di due genitori che possiedono entrambi lo stesso gene mutato, cioè sono portatori sani. E quindi si può notare che questo accade soltanto in quei casi in cui si verifica una serie di coincidenze genetiche; pertanto la malattia risulta essere estremamente rare. Molte malattie genetiche rare del sistema nervoso centrale hanno in comune questo aspetto dell’ereditarietà, si pensi alla SMA (atrofia muscolare spinale) o all’Atassia di Friedrich.
Il corso della RTD può avere inizio in qualsiasi momento, dalla nascita fino ai 30 anni di età. Nel mio caso, per fortuna, la malattia è insorta, in modo pieno, solo a cavallo tra l’infanzia e l’adolescenza, quando avevo 11/12 anni.. Già dapprima avevo avvertito qualche lieve sintomo, come la paresi facciale e qualche problema di deglutizione e di equilibrio nella camminata, che tuttavia non mi avevano causato grossi problemi, anzi, erano quasi del tutto scomparsi a seguito del trattamento con immunoglobulina.
Il periodo cruciale arrivò all’età di 11 anni, nel 2010, anno in cui la malattia raggiunse il suo stato più avanzato. Iniziai a notare che non avevo più la stessa agilità di prima, quando giocavo a calcio con gli altri bambini non riuscivo più a essere abile come lo ero prima, ma ovviamente ci riprovavo sempre, con l’ingenuità tipica dei bambini. Non sarei mai riuscito a spiegarmi una cosa del genere, e spesso insinuavo che forse avevo bisogno di un po’ di allenamento, e quindi i miei genitori, preoccupati, logicamente mi iscrissero a un centro di fisioterapia.
Le mie condizioni peggiorarono sempre di più, avevo quasi perso la capacità di camminare in maniera autonoma e avevo serie difficoltà a deglutire da cui, inevitabilmente, scaturivano problemi di alimentazione. La situazione divenne sempre più insostenibile, finché alla fine dovetti andare in ospedale perché andai incontro ad “ab ingestis” (cibo di traverso) a causa delle difficoltà di deglutizione, con tutte le conseguenze del caso.
In seguito, fui trasferito all’ospedale Pediatrico Bambin Gesù di Roma dove passai momenti molto drammatici. La mia vita era a rischio perché i medici non sapevano dove mettere le mani e non avevano la minima idea di quale potesse essere la mia diagnosi. La situazione era già di per sé tesa, ma a darle il colpo di grazia fu una crisi respiratoria che mi costò due mesi in rianimazione, la ventilazione artificiale e la tracheostomia.
Tuttavia, la storia ebbe un buon esito finale. Qualche mese prima che le mie condizioni degenerassero, era stato pubblicato quello che può essere considerato il primo, vero e proprio, studio sull’RTD, in cui si parlava degli effetti positivi del trattamento con la riboflavina. Di qui ebbe origine la fondamentale intuizione di un medico, esperto di malattie neuromuscolari, che associò le mie condizioni a quelle di un potenziale caso di RTD (al tempo BVVL, Brown Vialetto Van Laere) dando inizio al trattamento con riboflavina. Da quel momento in poi le mie condizioni iniziarono a migliorare grazie al trattamento con la riboflavina, e successivamente, per togliere ogni dubbio, la presenza dell’RTD venne geneticamente confermata.
Le mie condizioni migliorarono al punto che non ebbi più bisogno della ventilazione artificiale e il mio stato fisico mi consentì di affrontare un breve periodo di riabilitazione, al termine del quale ricevetti il via libera per tornare a casa.
Tuttavia, nonostante i grandi progressi iniziali, il mio stato clinico si è stabilizzato su livelli accettabili, ma non sufficienti.
Bisogna considerare, che nonostante la risposta favorevole alla riboflavina nella stragrande maggioranza degli individui con RTD (me compreso), essa non rappresenta una cura per la patologia. Il trattamento con riboflavina è in grado di stabilizzare/rallentare il processo della malattia, ma da sola non è sufficiente, e quindi vi è straordinario bisogno di aprire la strada verso altre strategie terapeutiche più efficaci.
Per quel che mi riguarda, in questi 9 anni che sono trascorsi dall’inizio del trattamento, la riboflavina mi ha garantito una buona qualità della vita, la stabilizzazione dello stato clinico su parametri accettabili, accompagnata da qualche lieve miglioramento, e soprattutto ha evitato quasi del tutto che insorgessero nuovi sintomi e che quelli esistenti deteriorassero.
Tuttavia, l’insieme dei sintomi da RTD, probabilmente di tipo 3 (sicché non ho grossi problemi di vista o di sordità), è rimasto essenzialmente intatto: la paralisi ponto-bulbare a causa della quale non riesco a muovere la maggior parte dei muscoli facciali, né a sorridere, chiudere le labbra, sbattere le palpebre o masticare senza affaticarmi; l’atrofia e le difficolta di movimento della lingua che significano problemi di deglutizione, e quindi, di alimentazione orale; la mancanza di equilibrio che mi impedisce di camminare in modo autonomo; il tono della voce che è totalmente assente e non mi consente di comunicare in modo opportuno; e così via. Di questo passo potrei continuare all’infinito, ma credo sia già abbastanza per comprendere quanto la vita possa essere difficile per coloro che debbono vivere con patologie neurologiche come la RTD.
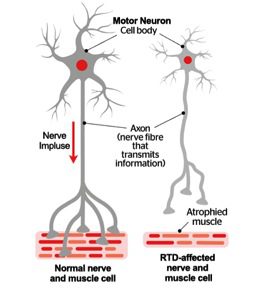
Tutti questi sintomi hanno origine dal fatto che la RTD è una patologia che colpisce le cellule nervose motorie e sensoriali (neuroni) del midollo spinale e del tronco encefalico. La regione del tronco encefalico più colpita è conosciuta come regione ponto-bulbare, e contiene sia neuroni motori che neuroni sensoriali, detti neuroni cranici.
In presenza della RTD si verifica un grave deterioramento, o addirittura, la perdita dei motoneuroni., e inevitabilmente ciò comporta uno stato di progressiva debolezza e atrofia (riduzione del volume) nei muscoli, inclusi quei muscoli responsabili del movimento, della respirazione e della deglutizione.
 Il futuro delle persone con RTD è incerto, le conoscenze attuali non consentono di fare previsioni a lungo termine. Tuttavia le ragioni per credere che tutto vada per il meglio sono davvero ben fondate, basti pensare al fatto che solo dieci anni fa non esisteva alcun trattamento comprovato in grado di arrestarne i sintomi. I progressi compiuti dalla scienza, in questo caso dalla scienza medica, rappresentano delle ottime ragioni per credere che un giorno la RTD, così come altre malattie genetiche del motoneurone, possano essere curate in modo soddisfacente.
Il futuro delle persone con RTD è incerto, le conoscenze attuali non consentono di fare previsioni a lungo termine. Tuttavia le ragioni per credere che tutto vada per il meglio sono davvero ben fondate, basti pensare al fatto che solo dieci anni fa non esisteva alcun trattamento comprovato in grado di arrestarne i sintomi. I progressi compiuti dalla scienza, in questo caso dalla scienza medica, rappresentano delle ottime ragioni per credere che un giorno la RTD, così come altre malattie genetiche del motoneurone, possano essere curate in modo soddisfacente.
I risultati raggiunti dalla moderna scienza fino a pochi anni fa rappresentavano solo delle prospettive molto difficili da raggiungere, dal sapore quasi fantascientifico. Oggi, invece, la scienza mette a disposizione una vasta serie di possibilità terapeutiche in grado di prolungare e migliorare la qualità della vita delle persone con malattie neurologiche, basti pensare ai grandi risultati ottenuti dalla terapia genica.
La nostra vita e le nostre speranze sono interamente riposte nelle mani della scienza. Abbiamo disperato bisogno di approfondire le conoscenze relative alla RTD e sviluppare nuove potenziali strategie terapeutiche da combinare con la riboflavina. Pertanto vi è la straordinaria necessità di sostenere nuovi studi e finanziare ulteriori ricerche.
Ma esiste un grande ostacolo di natura economica, dovuto al fatto che nessuno, né gli investimenti pubblici né quelli privati, hanno il minimo interesse a finanziare lo studio di una malattia per cui esistono così pochi casi. La fondazione Cure RTD e l’apparato solidale da cui è sostenuta, rappresentano, pertanto, un’ancora di salvezza attraverso cui vengono costantemente finanziati e sostenuti nuovi studi.
ABBIAMO BISOGNO DI VOI E DELLA VOSTRA SOLIDARIETA’!
Per maggiori informazioni consultare il sito: http://curertd.org/ (facilmente traducibile dall’inglese grazie alla traduzione automatica di google).
Per collaborare o fare una donazione, consultare l’apposita pagina del sito: http://curertd.org/waystohelp/
In alternativa: è possibile donare attraverso la pagina Facebook “Cure RTD”.



©Riproduzione riservata
